OHM: Allosteric Pathways — Two Perspectives
Results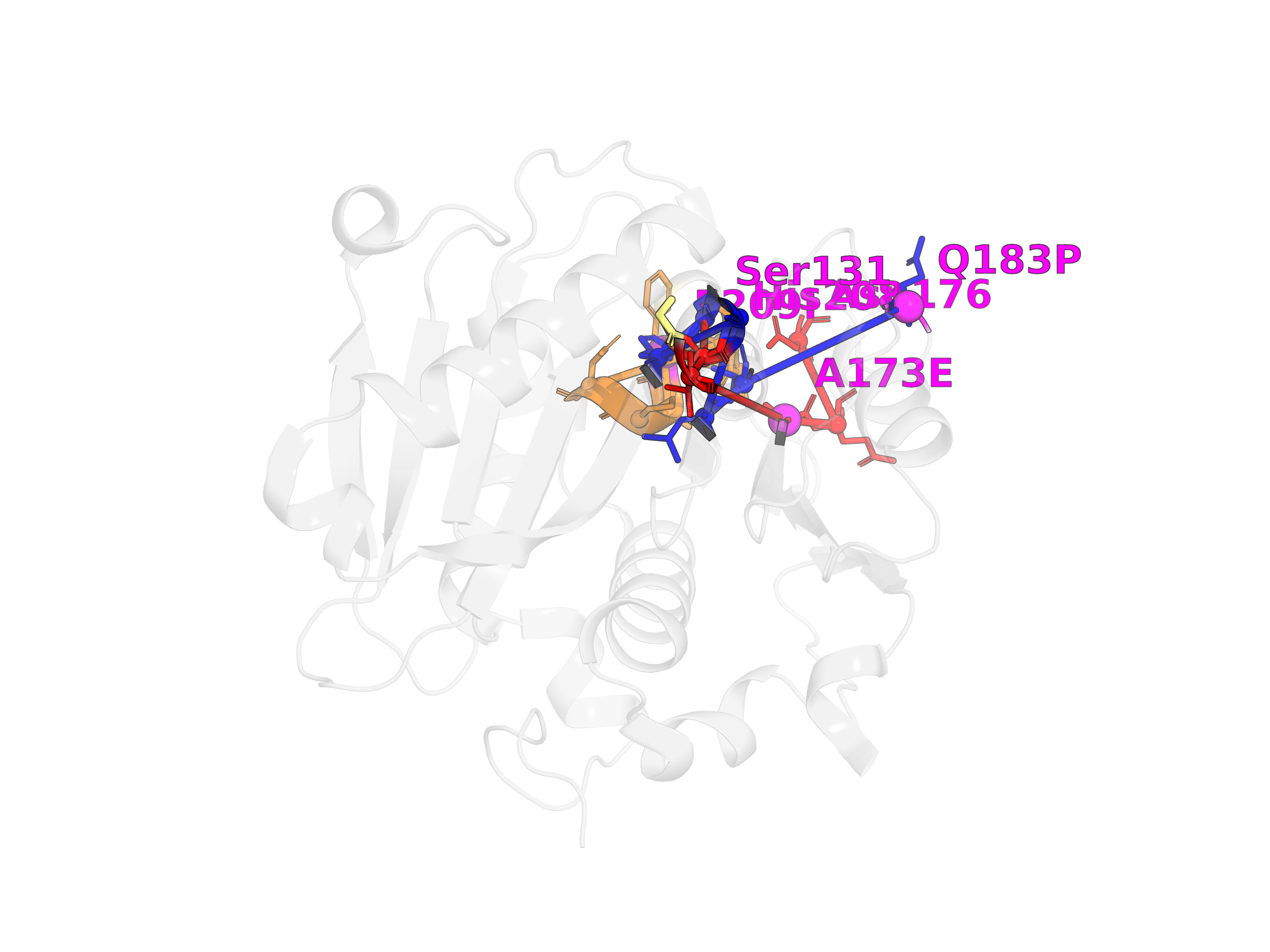
Red = Path 1 (core relay), Orange = Path 2 (substrate access), Blue = Path 3 (distal handle). Yellow = catalytic triad. Magenta = candidate mutation sites.
Path 1: Core Catalytic Relay · ACI > 95%
His164→Ser165(cat.)→Gly168→Thr171→Ala207→Glu208→Asp210(cat.)
A207E sits on this path — highest ACI (98.4%) among beneficial mutations. Directly connects two catalytic residues.
Path 2: Substrate Access · ACI > 90%
Ser241→Phe243→Ala244→Pro245→Asn246(S+1)→His242(cat.)
ICCG's F243I lies on this path — explains why this deleterious single is epistatically rescued in the ICCG combination.
Path 3: Distal Handle · ACI 85-95%
Gln217→Thr188→Leu187→Gly169→[junction]→Ser165(cat.)
Q217P = allosteric handle, rigidifies loop. Longest-range path (~25 Å surface→active site).
From Ser165 (Nucleophile) Outward
ACI radiates outward through a highly conserved, mutationally intolerant core:
| Pos | AA | ACI% | Cons | Fitness | Insight |
|---|---|---|---|---|---|
| 164 | His | 99.6 | 1.00 | −0.18 | DO NOT TOUCH — relay backbone |
| 168 | Gly | 99.2 | 1.00 | −0.13 | DO NOT TOUCH — relay backbone |
| 167 | Gly | 98.1 | 1.00 | −0.67 | GxSxG motif (i+2 after Ser165) — IMMUTABLE |
| 171 | Thr | 96.9 | 0.84 | +0.03 | Neutral — tolerable but no gain |
| 169 | Gly | 95.0 | 1.00 | −0.42 | Path 3 junction — Q217P relay feeds through here |
| 166 | Met | 93.8 | 1.00 | −0.47 | Conserved relay residue |
| 170 | Gly | 92.2 | 0.97 | −0.47 | Relay backbone |
| 163 | Gly | 91.9 | 1.00 | −0.46 | Relay backbone |
Insight: The Ser165 relay core is entirely locked by conservation + negative fitness. Engineering must approach from the periphery (Path 3: Q217P → ... → Ser165).
From Asp210 (Acid) Outward
| Pos | AA | ACI% | Cons | Fitness | Insight |
|---|---|---|---|---|---|
| 207 | Ala | 98.4 | 0.85 | +0.23 | ★ A207E — HOTSPOT on relay! |
| 209 | Ala | 96.1 | 0.46 | +0.04 | Low conservation — handle zone |
| 213 | Ala | 94.6 | 1.00 | +0.13 | Synergistic (A213T+T230A Δ=+1.22) |
| 212 | Val | 94.2 | 0.84 | −0.02 | Neutral |
| 208 | Glu | 91.1 | 0.90 | −0.05 | Conserved, relay core |
| 217 | Gln | 84.9 | 0.53 | +0.34 | ★ Q217P — HANDLE + HOTSPOT |
Insight: Position 207 is the only high-ACI position near Asp210 that is also a DMS hotspot. All other high-ACI neighbors are conserved + deleterious.
From His242 (Base) Outward
| Pos | AA | ACI% | Cons | Fitness | Insight |
|---|---|---|---|---|---|
| 241 | Ser | 98.8 | 0.80 | −0.38 | Handle — substrate entry |
| 244 | Ala | 97.3 | 0.75 | −0.10 | Handle zone |
| 240 | Ala | 96.5 | 0.99 | −0.27 | Core relay, conserved |
| 245 | Pro | 95.7 | 1.00 | +0.15 | Only mutable residue on channel |
| 243 | Phe | 93.4 | 0.75 | −0.29 | ICCG F243I sits here |
| 246 | Asn | 93.0 | 0.99 | −0.75 | Binding pocket (subsite +1) |
| 238 | Asp | 90.3 | 0.55 | −0.20 | ICCG D238C disulfide partner |
Insight: The His242 channel is tightly optimized. ICCG mutated F243I in this channel (individually deleterious but combinatorially rescued by the other 3 ICCG mutations).